
Epigenetic regulation of cancer driving pathways (emQTL)
Identification of epigenetically regulated pathways in cancer is possible using the expression methylation quantitative trait loci (emQTL) approach published in Fleischer, Tekpli et al., Nat Commun 2017. Using an improved version of this method, we have now identified epigenetic dysregulation associated with increased activity of the cell cycle, showing that epigenetic dysregulation in triple negative breast cancer may provide a growth advantage in these tumors.
By grouping emQTLs by biclustering we identify associations representing important biological processes associated with breast cancer pathogenesis such as proliferation and tumor infiltrating fibroblasts. We observe hypomethylation at enhancers carrying transcription factor binding sites of key proliferation-driving transcription factors such as CEBP-β, FOSL1, and FOSL2, with concomitant high expression of cell cycle- and proliferation-related genes in aggressive breast tumors. This loss of methylation is especially clear in ER negative tumors, but the trend is apparent also within ER positive tumors suggesting that epigentic regulation of proliferation may occur in all subtypes [Ankill et al., NAR Cancer 2022]
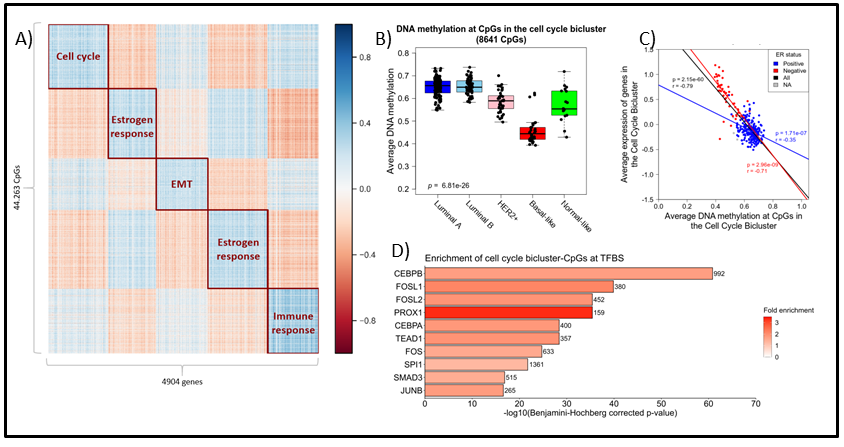
Figure 1: A) Heatmap displaying the five biclusters identified by spectral co-clustering when k (number of clusters) was set to 5. Columns represents genes (n = 4904) and rows represents CpGs (n = 44,263). Blue points indicate strong negative correlations between the variables while red points represent positive correlations. White points indicate little or no correlation. B) ) Boxplot showing the average DNA methylation of the cell cycle bicluster-CpGs by PAM50 subtype. Kruskal-Wallis test p-values are denoted in the lower left corner. C) Scatterplot showing the association between average DNA methylation and average gene expression in the cell cycle bicluster. ER status denoted by blue (ER positive) and red (ER negative). Pearson correlation coefficients and p-values are denoted and colored by ER status. D) Bar plot representing enrichment of the cell cycle bicluster-CpGs at the binding site of specific TFs according to UniBind [Gheorghe et al., NAR 2019]. Bar length display the log-transformed BH-corrected p-value obtained by hypergeometric testing for each TF. Color indicates fold enricment.
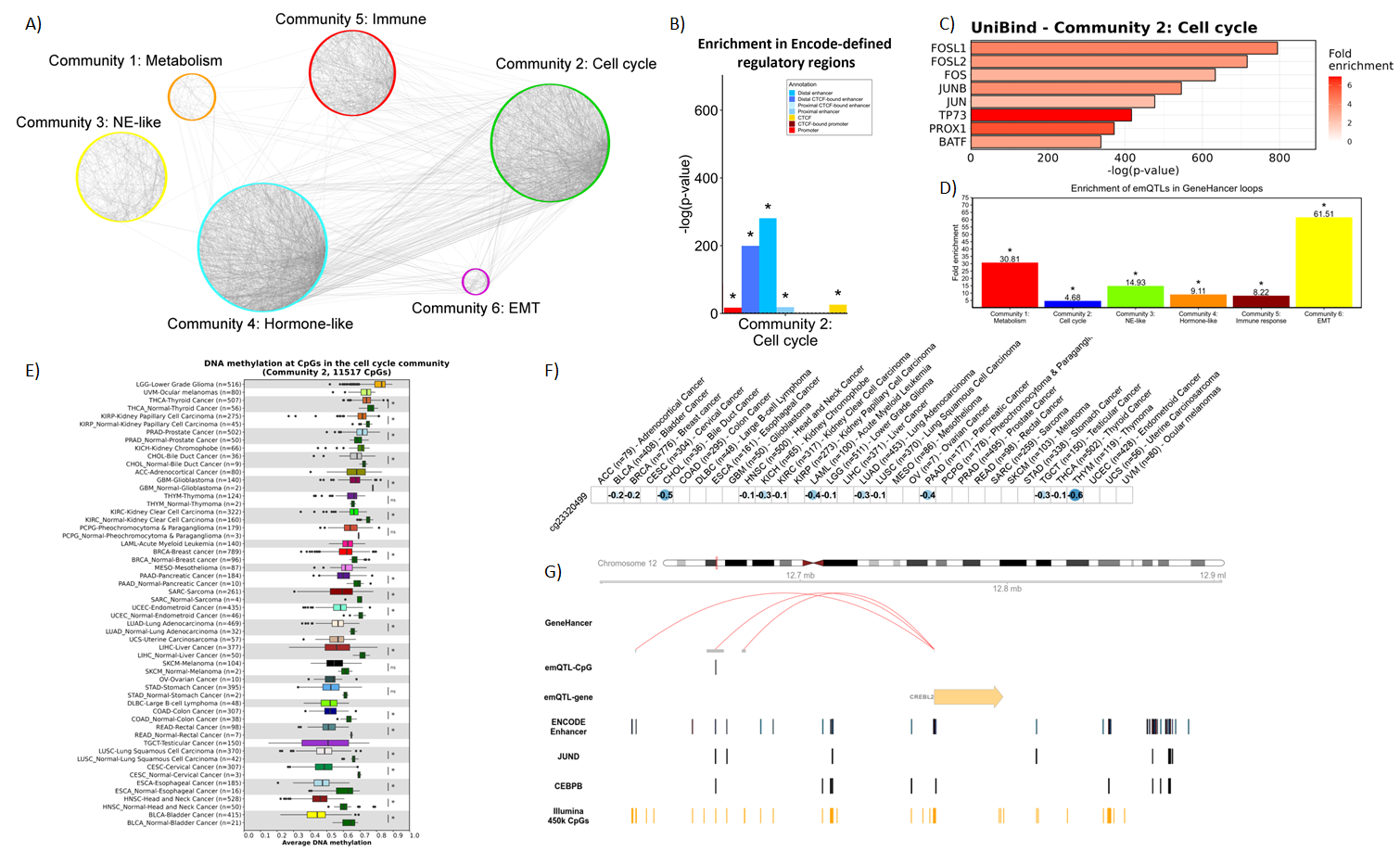
Following the results from breast cancer, we are now applying the method pan-cancer. Using data from more than 30 cancer types, we observe that proliferation is linked to loss of methylation across most cancer types, and that the loss of methylation occurs in regulatory regions and binding sites of several proliferation-related trancription factors.
Participants: