
CRISPR epigenetic editing
The aim of this project is to assess the functional and causal effects of the identified epigenetic alterations identified through the emQTL approach. To identify the top candidates, we're using a supervised version of the emQTL approach, where candidate CpG-gene pairs need fulfill several criteria. 1) a CpG needs to be in an enhancer region, 2) a CpG needs to be in the transcription factor (TF) binding region of a TF of interest, 3) a CpG-gene pair must be on opposite sides of a chromatin loop, and 4) a gene needs to be part of a gene set of interest (see Figure 1 for example).
To functionally assess the importance of the indentfied candidates, we are using CRISPR epigenetic editing. In breast cancer cell lines with stable and inducible expression of deactivated Cas9 fused with the catalytic domain of DNMT3A, we can induce DNA methylation at specific loci. The effects of induced DNA methylation is assessed by measuring transcription factor occupancy, target gene expression, and cell proliferation and migration.
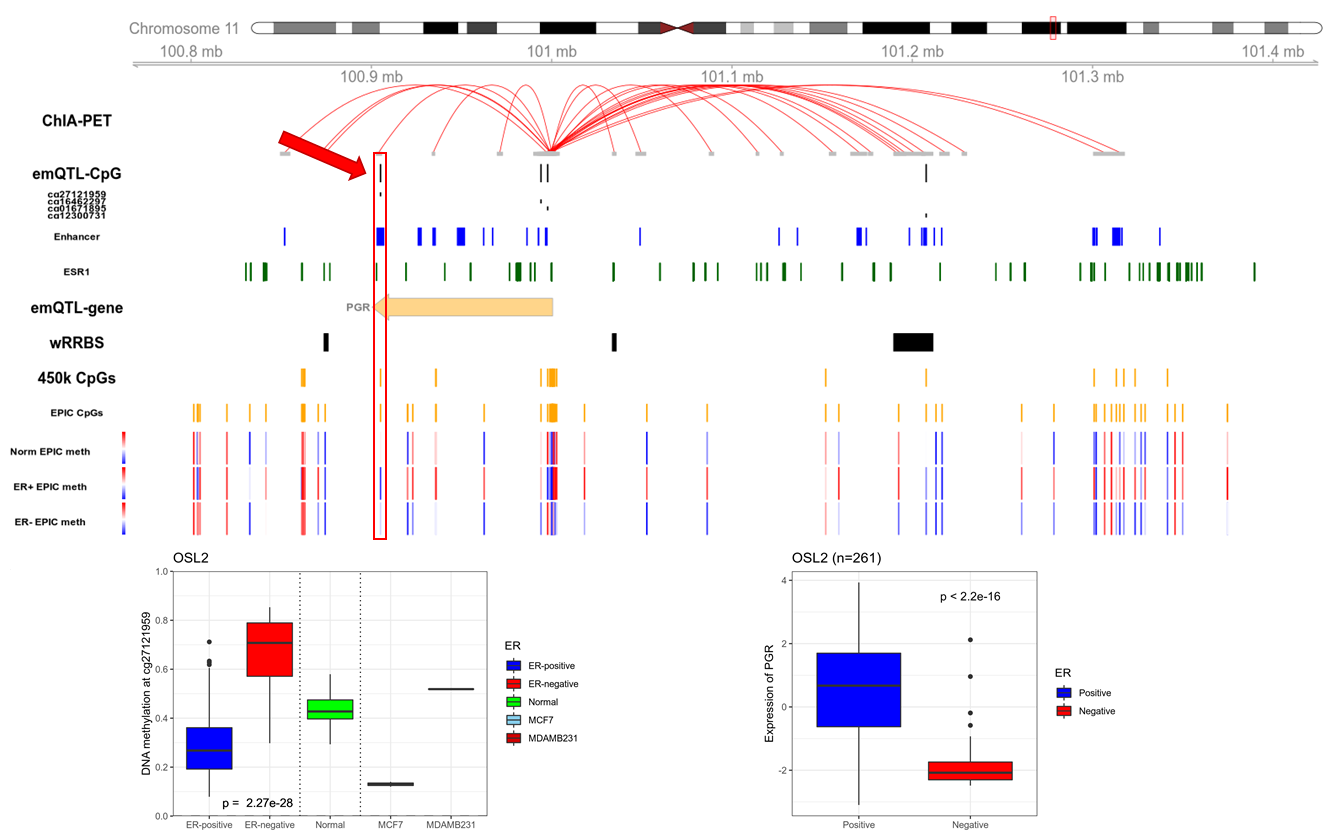
Figure 1: Candidate emQTL CpG-gene pair. The CpG cg27121959 is located in an enhancer with an ER binding site, in the 3'UTR of the PGR gene. The CpG is connected to the transcription start site of PGR through a chromatin loop. ER positive tumors have low methylation while ER negative tumors have high methylation, implying that this enhancer is active in ER positive disease. Potential causal regulation by the methylation of this enhancer will be studied by inducing methylation.
Participants: