
Studying the effect of the Stormorken syndrome causing mutation STIM1 R304W in mouse models
Repeated bleeding in a young boy was how the first patient came to Professor Helge Stormorken’s attention in 1974. Patients with Stormorken syndrome (OMIM 185070) exhibit mild bleeding tendency, thrombocytopathy, thrombocytopenia, mild anemia, splenic aplasia/functional asplenia, tubular aggregate myopathy, and miosis (Stormorken et al., 1985; Stormorken, 2002).
We have shown that the missense mutation STIM1 p.R304W causes the autosomal dominant Stormorken syndrome in four families (Misceo et al., 2014). In a parallel study, the same mutation was identified in two additional families (Nesin et al., 2014). The R304W substitution is the most common STIM1 mutation, so far identified in 18 patients with Stormorken syndrome in 13 families of different geographic origin.
STIM1 is an endoplasmic reticulum (ER) membrane protein that initiates store-operated Ca2+ entry (SOCE) into the cell when ER Ca2+ levels are low. ER Ca2+ store depletion activates STIM1 by releasing an intramolecular "clamp" formed between two coiled coil domains (CC1 and CC3) of the protein, enabling the C terminus to extend and interact with the plasma membrane protein Orai1. Orai1 is a Ca2+ release-activated calcium (CRAC) channel that enables Ca2+ influx into the cytoplasm.
In order to study the physiological consequences of the STIM1 R304W mutation, we established knock-in mice expressing this mutation. STIM1 R304W was lethal in the homozygous state, however, the few surviving homozygous mice and the heterozygotes presented with skeletal muscle degeneration (Figure 1; Gamage et al., 2018). The mice also presented with increased bone volume, as well as subgingival hair growth on the lower incisors (Figure 1; Gamage et al., 2020).
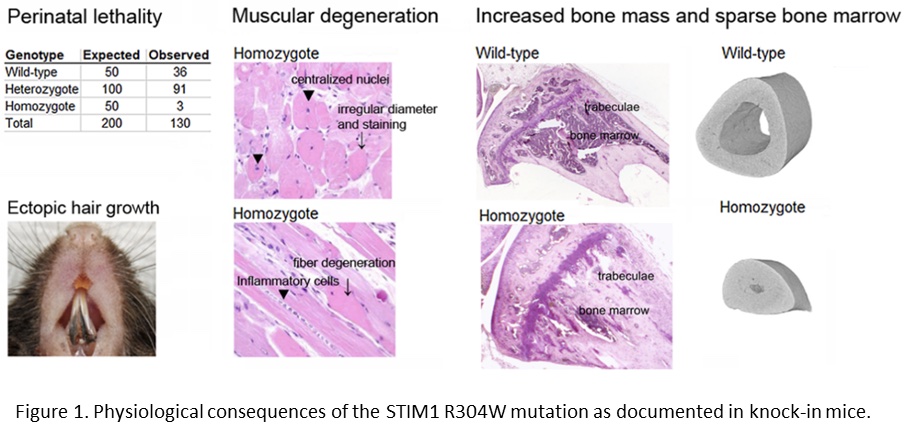
The findings in our mouse model highlight the importance of STIM1 in the development and homeostasis of the skeleton and the skeletal muscle. In mice, STIM1 R304W also seems to induce an abnormal epithelial cell fate.
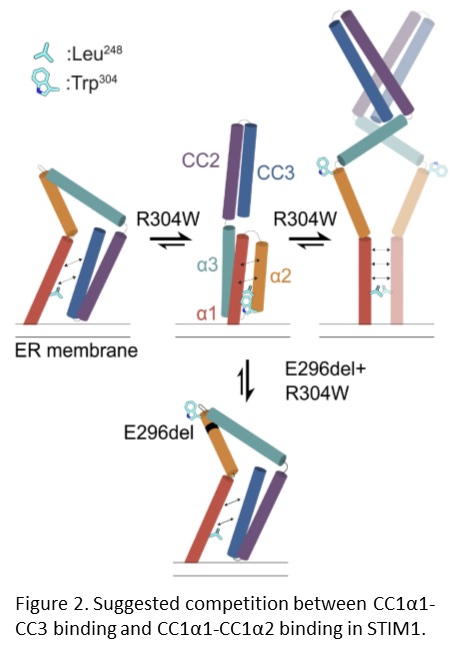
Our collaborators previously showed that the STIM1 R304W mutation destabilizes the resting state conformation of STIM1, resulting in a constitutively extended and therefore active STIM1, which leads to constitutive Ca2+ influx into the cell (Fahrner et al., 2018). We further documented that double mutant mice expressing STIM1 R304W and the deletion of Glu296 in cis, showed completely reversal of the pathophysiological effects of the STIM1 R304W mutation (Gamage et al., 2023). NMR spectroscopy, molecular dynamics simulations and live-cell experiments revealed that the deletion of Glu296 reversed the extended conformation of STIM1 R304W. Thus, the R304W and Glu296 double mutation restored the STIM1 CC domain interactions that are critical for proper regulation of the CRAC channel (Figure 2).
National collaborators:
Prof. G. Gunnes (NMBU): Pathology and histology
Dr. W. Louch and Prof. G. Christiansen (OUS): Ca2+ influx analyses
Profs. P.A. Holme and G. Tjønnfjord (OUS): Hematology
Profs. S.P. Lyngstadaas and J.E. Reseland (UiO): Analysis of bone structure
International collaborators:
Prof. C. Romanin (Johannes Kepler University Linz, Austria): STIM1 conformational changes
Prof. W. Bergmeier (University of North Carolina at Chapel Hill, USA): Mouse platelet biology
Prof. R. Lacruz (Sackler Institute, NYU School of Medicine, USA): Tooth development
Our papers in this project:
- Misceo et al. (2014). Human Mutation 35:556-564.
- Gamage et al. (2018). Cell Calcium 76:87-100.
- Gamage et al. (2020). Cell Calcium 85:102110.
- Gamage et al. (2023). Science Signaling 16(771):1-17.