
Welcome to Doriana Misceo’s project group: Novel genes causing primary cilia dysfunction in congenital brain malformations

Project group leader
Photo: Bård Gudim AS
The aim of the project is to identify genetic and molecular basis of congenital brain malformation caused by dysfunction of primary cilia. Nearly all cells in the human body can build a primary cilium to sense the external environment for biochemical, physical and mechanical stimuli and trigger appropriate cell responses. Cilia are necessary for a wide range of physiological processes already during embryonic life. Structural or functional defects or insufficient number of primary (and motile) cilia can result in isolated or multiple organ abnormalities, called ciliopathies. These are rare or ultra-rare diseases remarkable for the phenotypic and genetic heterogeneity and for the phenotypic overlaps. In the brain, primary cilia play crucial roles for development, neuronal connectivity and survival. Ciliary dysfunction is associated with a wide range of congenital brain malformations, comprising mid-hindbrain malformation, migration disorders or the absence of major brain structures. In addition, cilia dysfunction has been linked to the major psychiatric disorders.
More than 200 ciliopathies have been defined clinically and genetically, however more than 50% of patients with ciliopathies remain without a genetic diagnosis after diagnostic WES or WGS analyses.
Recently, we reported three unrelated patients, one recruited by Dr. Inger-Lise Mero (AMG) and two identified through GeneMatcher (genematcher.org), with clinical diagnosis of Strømme syndrome. This ultra-rare primary ciliopathy is caused by bi-allelic pathogenic variants in CENPF, which encodes a microtubule regulating protein. In one of the patients, we used Long Reads-Whole Genome Sequencing (LR-WGS) (PacBio approach) to identify a 5323 bp deletion overlapping CENPF exon 1 (Figure 1). This is the first report of a CENPF intragenic deletion in a patient with Strømme syndrome. CENPF exon 1 is flanked by sequence repetitive elements and may therefore represent a site of a recurrent structural variation. We suggest that this genomic region should be carefully investigated in patients with a clinical diagnosis of Strømme syndrome, lacking genetic finding after SNV/Indels analysis.
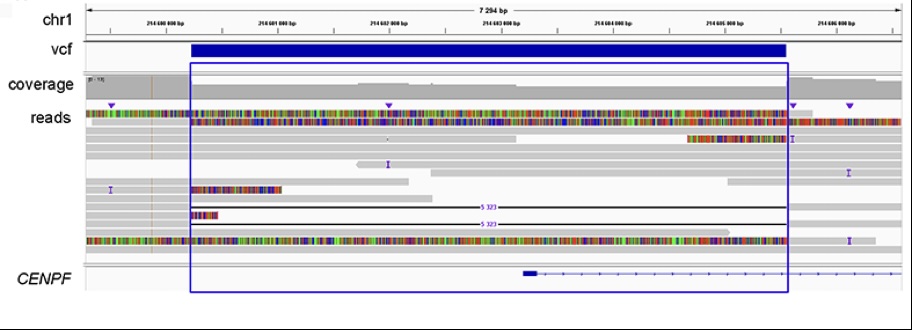
Patients with Strømme syndrome usually present in the newborn period with intestinal atresia, microcephaly, and eye anomalies. Even though bi-allelic CENPF null variants were detected in all the three patients in our report, clinical manifestations and severity varied remarkably. One patient presented with cerebral, ocular, intestinal and renal anomalies, which caused with neonatal lethality, while the other two were less severely affected, one of which had normal cognitive skills at 7 years of age. The genetic basis of this clinical variation motivates further research.
Our overall approach allows us to identify novel ciliopathy genes and disease mechanisms causing congenital brain malformations, study genotype-phenotype correlations and at the same time expand the experimental framework, which is important for the evaluation of variant pathogenicity and relevant for all genetic diseases.