Selected projects
Here we present a few selected project where the Core Facility has contributed with experimental structures, structural models or analyses, or experimental studies of interactions.
The current activity is high, with both exciting and novel structural biology projects and training of external users in our laboratory. Per 1.1.2025, the facility has assisted more than a hundred different research groups with a few hundred different projects, resulting in around 150 peer-review publications in international journals. More than 50 PhD studens have also recived services at our facility throughout our period of operation.
The projects range from smaller service functions to complete collaborative projects with the aim to determine novel structures of proteins/enzymes that are potential therapeutic targets, modeling of peptides and small molecules The completed and ongoing projects span diverse research fields such as metabolic diseases, cancer, immunology, heart and cardiovascular diseases, viral infections, regulation of transcription and replication, RNA modification and DNA repair.
Examples
Autoantibody binding and unique enzyme-substrate intermediate conformation of human transglutaminase 3 - The enzyme TG3 targets glutamine residues in polypeptides, and modifies them by transamidation or deamidation. In this study, Heggelund and co-workers used X-ray structures of inactive and active human TG3 in complex with a TG3-specific antibody fragment from a dermtitis herpetiformis patient. These analyses support a model for how receptors on B-cells of the immune system bind and internalize TG3-gluten complexes, which in turn caues immune cells to present antigens against gluten.
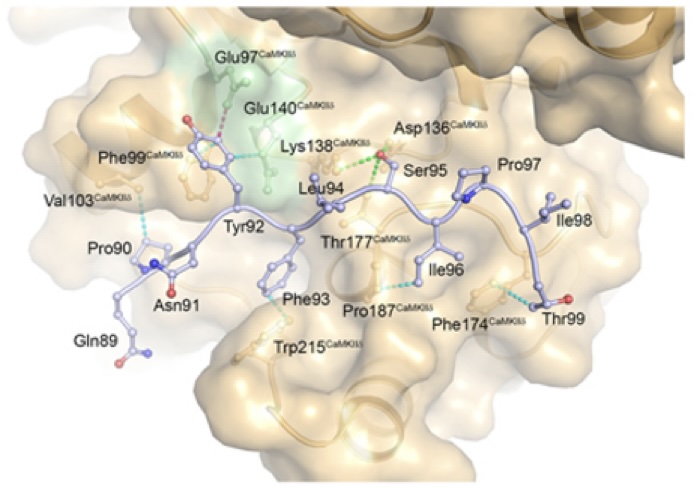
AKAP18δ Anchors and Regulates CaMKII Activity at Phospholamban-SERCA2 and RYR - In this stydy, Carlson and co-workers show that the protein AKAP18δ uses two different regions in its sequence to regluate the activity of the kinase CAMKIIδ when in complex with SERCA2-PLN and RYR. The SERCA2-PLN and RYR proteins are responsible for relaxation and contraction of cardiomyocytes - the cells that are responsible for contractile force in the heart.
An engineered human albumin enhances half-life and transmucosal delivery when fused to protein-based biologics - In this study, Bern et. al., describes a mutant (QMP) human serum albumin (HSA) variant with three mutations that improves the binding of HSA to the neonatal Fc receptor (FcRN) expressed on mucosal epithelial cells. The QMP-HSA mutant binds to FcRN and is protected from degradation. This, in turn, allowed better uptake and prolonged half-life of proteins fused to QMP-HSA when these were administered by intranasal delivery in a mouse model. The delivery of protein drugs via the mucosal pathway by use of a nasal spray is much more convenient for the patient, than the traditional intravenous or subcutaneously (under the skin) methods, however the method is limited by transport over the mucosal barrier. The QMP-HSA seems to be a good tool to overcome this problem with poor uptake over the mucosal barrier. The study is published in Science Translational Medicine (2020), 12:565, and the design of the QMP mutants based on structural analysis of the HSA-FcRN complex.
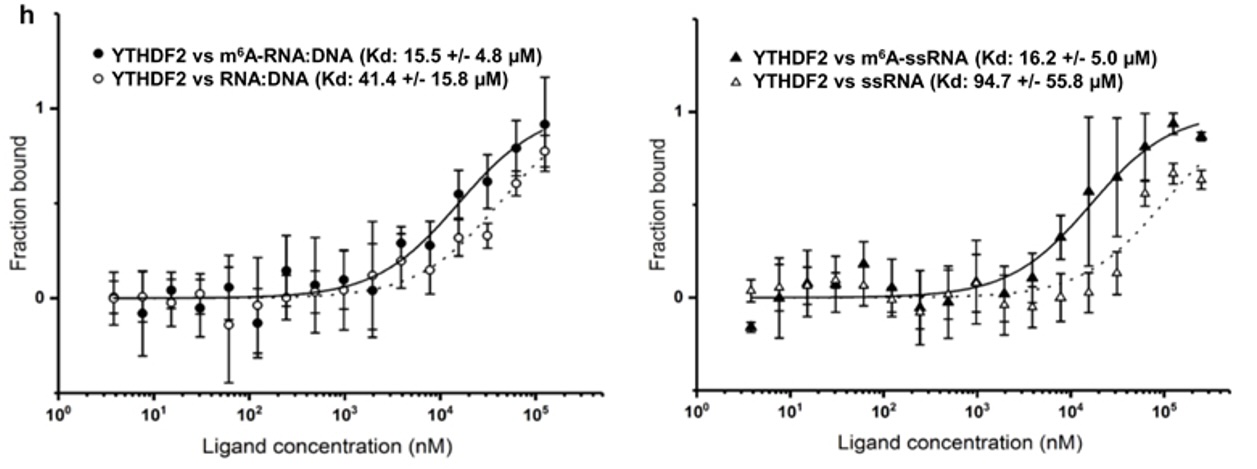
N6-methyladenosine regulates the stability of RNA:DNA hybrids in human cells -In this study, published in Nature Genetics, Abakir et. al. show that the epigenetic marker N6-methyladenosine (m6A) - a factor contributing to diffferent aspects of messenger RNA metabolism - is detectable in a majority of RNA:DNA hybrids. When m6A is present in R-loops, a structure formed by RNA:DNA hybrids, the m6A 'reader' protein YTHDF2 binds to these structures and is involved in the degradation of mRNA. The interaction between R-loops, with and without m6A, and YTHDF2 were confirmed by microscale thermophoresis (MST).
Axitinib blocks Wnt/beta-catenin signaling and directs asymmetric cell division in cancer. - In this study by Yi Qu, Karl-Henning Kalland, Xisong Ke and co-workers, it is demonstrated that the clinically apporved drug axitinib blocks Wnt/β-catenin signaling in cancer cells. Our facility contributed with experiments to show a direct interaction between the axitinib drug and the the E3 ubiquitin ligase SHPRH as the drug target.
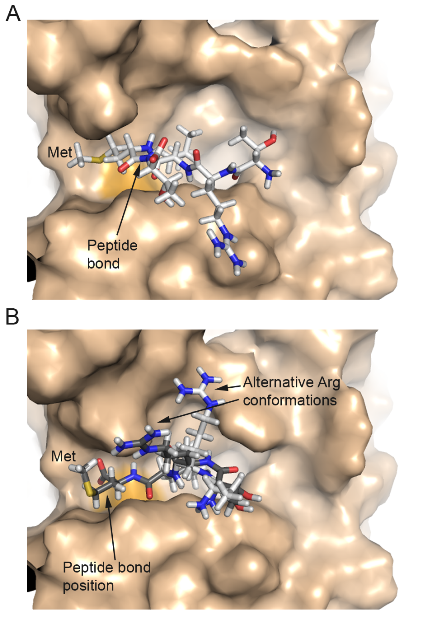
Protein Phosphatase 1c Associated with the Cardiac Sodium Calcium Exchanger 1 Regulates Its Activity by Dephosphorylating Serine 68-phosphorylated Phospholemman - The sodium-calcium Ca exchanger 1 (NCX1) is an important regulator of intracellular calcium homeostasis. In this study, Hafver, Sejersted, Calsson and co-workers show that the catalytic subunit of protein phosphatatse 1 (PP1) might interact directly with the NCX1 receptor. The study also includes a model for the interaction between PP1 and NCX1.
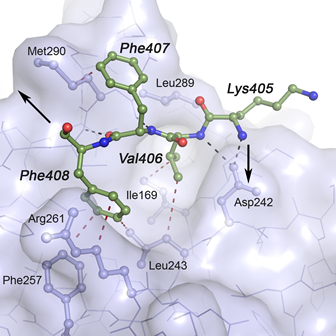
Mutation analysis in Norwegian families with hereditary hemorrhagic telangiectasia: founder mutations in ACVRL1 - In this study, Heimdal, Kuseth and co-workers present an analysis of mutations in the gene ACVRL1 found in families with patients suffering from HHT (Osler-Weber-Rendu disease) which leads to epistaxis and mucocutaneous telangiectasias and arteriovenous malformations (AVMs) in internal organs. We contributed with an anaysis of key mutants and their possible role in destabilization of the ACVRL1 protein.
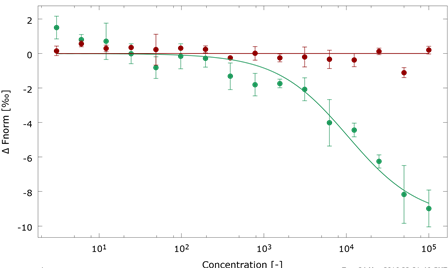
Modeling of molecular basis for calpain cleavage and inactivation of sodium-calcium exhanger 1 in heart failure - Cardiac sodium (Na+)-calcium (Ca2+) exchanger 1 (NCX1) is central to the maintenance of normal Ca2+ homeostasis and contraction. Studies indicate that the Ca2+-activated protease calpain cleaves NCX1. NCX1 protein along with calpain is up-regulated in aortic stenosis patients and rats with heart failure. Patients with coronary artery disease and sham-operated rats were used as controls. Calpain co-localized, co-fractionated, and co-immunoprecipitated with NCX1 in rat cardiomyocytes and left ventricle lysate. Immunoprecipitations, pull-down experiments, and extensive use of peptide arrays indicated that calpain domain III anchored to the first Ca2+ binding domain in NCX1, whereas the calpain catalytic region bound to the catenin-like domain in NCX1. The use of bioinformatics, mutational analyses, a substrate competitor peptide, and a specific NCX1-Met369 antibody identified a novel calpain cleavage site at Met369. Engineering NCX1-Met369 into a tobacco etch virus protease cleavage site revealed that specific cleavage at Met369inhibited NCX1 activity (both forward and reverse mode). Finally, a short peptide fragment containing the NCX1-Met369 cleavage site was modeled into the narrow active cleft of human calpain. Inhibition of NCX1 activity, such as we have observed here following calpain-induced NCX1 cleavage, might be beneficial in pathophysiological conditions where increased NCX1 activity contributes to cardiac dysfunction. Publication
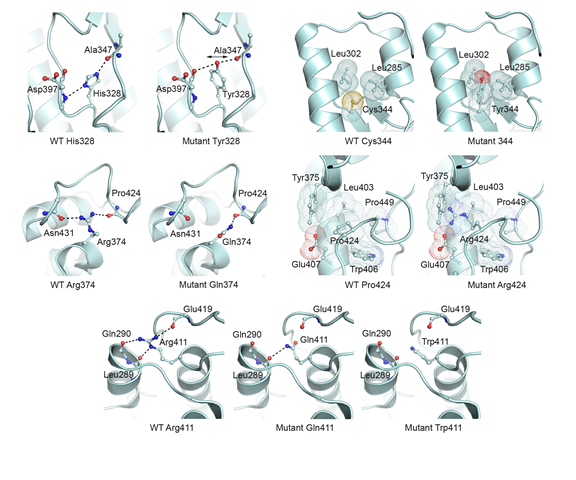
Modelling mutations in PGM3 - defects in glycosylation cause congenital disorder - The human gene PGM3 catalyzes the conversion of isomers of sugar molecules, and is key to many processes that depend on correct glycosylation. The paper presents data from patients suffering from recurrent infections, congenital leukonepia, B and T cell lympopenia and progression to bone marrow failure. The effect of disovered muatations in these patients in the PGM3 gene is discussed. Publication.
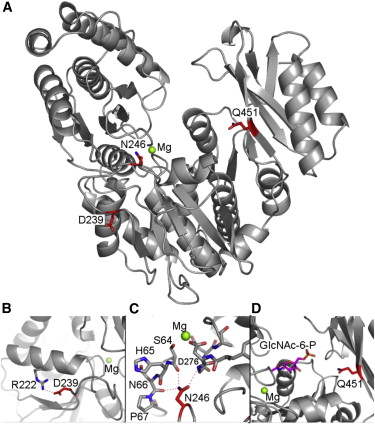
Transmembrane DinQ peptide modeling - The SOS regulated gene DinQ translates into a toxic single transmembrane spanning peptide localized in the inner membrane of E. coli. The DinQ peptide is extremely toxic, and for that reason highly regulated at different levels (translation, mRNA processing, expression). DinQ and similar tocix peptides are attractive models for antibacterial peptide drugs. Publication.
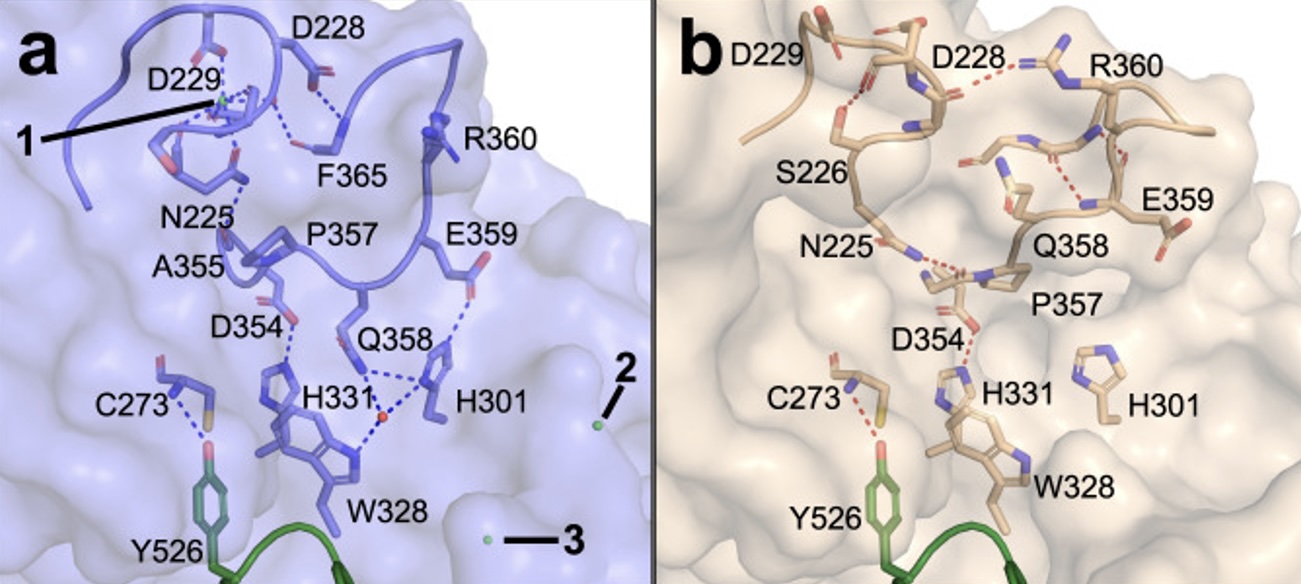
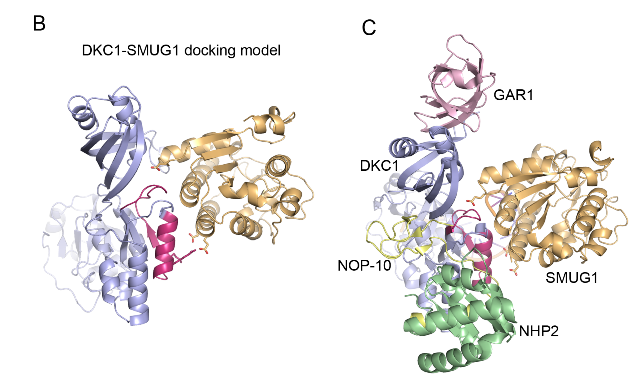
SMUG1-DKC1 complex involved in RNA quality control in human cells - Single-strand-selective monofunctional uracil-DNA glycosylase 1 (SMUG1), a base excision repair enzyme that removes uracil and oxidised pyrimidines from DNA, interact with the pseudouridine synthase Dyskerin (DKC1) and colocalizes with DKC1 in nucleoli and Cajal bodies. SMUG1 has activity on single-stranded RNA containing 5-hydroxymethyldeoxyuridine, and associates with the 47S rRNA precursor processed by DKC1. Depletion of SMUG1 leads to a reduction in the levels of mature rRNA, increase in polyadenylated rRNA, and the combined loss of SMUG1 and DKC1 leads to accumulation of 5-hydroxymethyluridine in rRNA. In conclusion, SMUG1 is a DKC1 interaction partner that contributes to rRNA quality control, partly by regulating 5-hydroxymethyluridine levels. A protein-protein docking model, supported by peptide array data, suggests the 3D structure of the SMUG1-DKC1 complex. Publication.
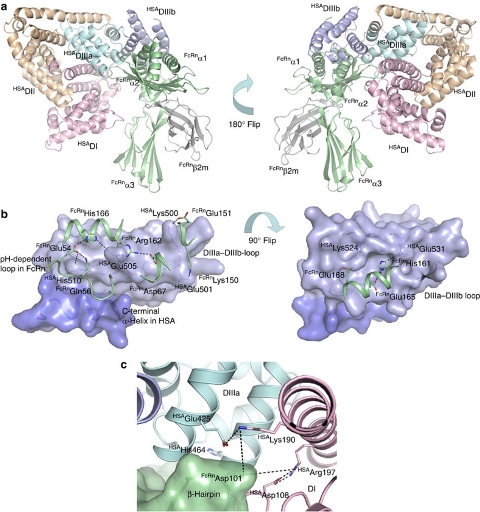
Complex between human serum albumin and the FcRn receptor controls albumin serum half-life - Albumin is the most abundant protein in blood where it has a pivotal role as a transporter of fatty acids and drugs. Albumin has long serum half-life, protected from degradation by pH-dependent recycling mediated by interaction with the neonatal Fc receptor, FcRn. Here we present structure-based modelling of the FcRn-albumin complex, supported by binding analysis of site-specific mutants, providing mechanistic evidence for the presence of pH-sensitive ionic networks at the interaction interface. These networks involve conserved histidines in both FcRn and albumin domain III. Histidines also contribute to intramolecular interactions that stabilize the otherwise flexible loops at both the interacting surfaces. Molecular details of the FcRn-albumin complex may guide the development of novel albumin variants with altered serum half-life as carriers of drugs. Publication.
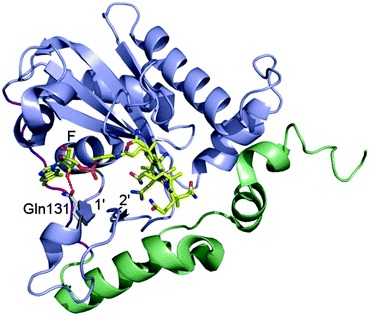
Mutation in MMACHC gene leads to late onset disease - Combined methylmalonic aciduria and homocystinuria, cblC type (MMACHC), is the most common inborn error of cellular vitamin B12 metabolism and is caused by mutations in the MMACHC gene. This metabolic disease results in impaired intracellular synthesis of adenosylcobalamin and methylcobalamin, coenzymes for the methylmalonyl-CoA mutase and methionine synthase enzymes, respectively. The inability to produce normal levels of these two coenzymes leads to increased concentrations of methylmalonic acid and homocysteine in plasma and urine, together with normal or decreased concentration of methionine in plasma. Here, we report a novel homozygous deletion mutation (NM_015506.2:c.392_394del) resulting in an in-frame deletion of amino acid Gln131 and late-onset disease in a 23-year-old male. The patient has sensory and motoric disabilities, urine and fecal incontinence, and light cognitive impairment. Studies on patient fibroblasts together with spectroscopic activity assays on recombinant MMACHC protein reveal that Gln131 is crucial in order to maintain enzyme activity. Furthermore, structural analyses show that Gln131 is one of only two residues making hydrogen bonds to the tail of cobalamin. Circular dichroism spectroscopy indicates that the 3D structure of the deletion mutant is folded but perturbed compared to the wild-type protein. Publication.
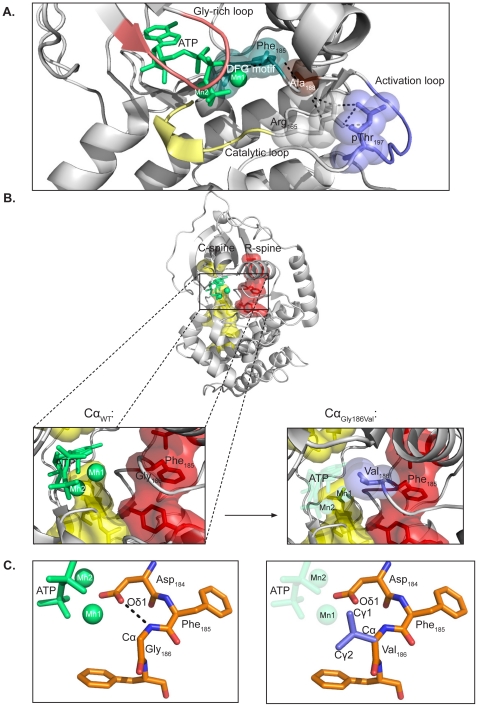
Identification and characterization of novel mutations in catalytic subunit of human protein kinase A - The genes PRKACA and PRKACB encode the principal catalytic (C) subunits of protein kinase A (PKA) Cα and Cβ, respectively. Cα is expressed in all eukaryotic tissues examined and studies of Cα knockout mice demonstrate a crucial role for Cα in normal physiology. Sequencing of PRKACA from 498 Norwegian donors and analysis of public databases identified four interesting nonsynonymous point mutations, Arg45Gln, Ser109Pro, Gly186Val, and Ser263Cys, in the Cα1 splice variant of the kinase. The different Cα variants were analyzed for kinase activity and regulatory (R) subunit binding. Mutation of Ser109 significantly reduced kinase activity while R subunit binding was unaltered. Mutation of Cα Gly186 completely abrogated kinase activity and PKA type I but not type II holoenzyme formation. Gly186 is located in the highly conserved DFG motif of Cα and mutation of this residue to Val is predicted to result in loss of binding of ATP and Mg2+, which may explain the kinetic inactivity. We hypothesize that individuals born with mutations of Ser109 or Gly186 may be faced with abnormal development and possibly severe disease. Publication.
Homology modelling of TGR5 - TGR5, the G protein-coupled bile acid receptor, has been linked to inflammatory pathways as well as bile homeostasis, and could therefore be involved in primary sclerosing cholangitis (PSC) a chronic inflammatory bile duct disease. In this project the TGR5 sequence variation in PSC patients were investigated. A series of mutations were identified, and a structural model of the TGR5 receptor was used to analyse the clinical data. Publication.
Analysis of human PCSK9 - Proprotein convertase subtilisin/kexin type 9 (PCSK9) binds to the low density lipoprotein receptor (LDLR) at the cell surface and disrupts the normal recycling of the LDLR, leading to LDLR degradation and reduced cellular uptake of plasma LDL. The facility has assisted in model building and analysis of the 3D structure of PCSK9 with respect to conserved regions involved in receptor interaction. The results provide important information for future studies of PCSK9 mutants and for investigations on the function of this regulator of cholesterol homeostasis. Publications: 1, 2, 3, 4, 5 and 6
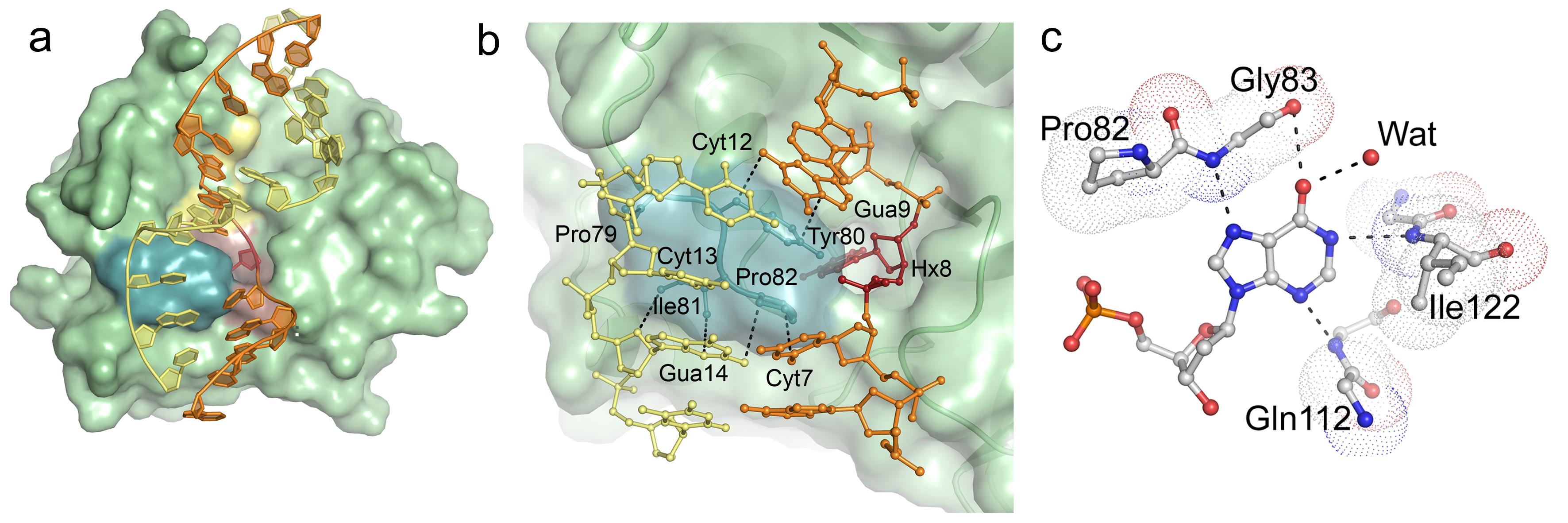
Structural basis for recognition of deaminated DNA by Endonuclease V - The DNA repair enzyme Endonuclease V recognizes deaminated adenine and guanine in DNA and initiate repair by cleaving the DNA strand to remove the damaged base. If left unrepaired, these deaminated bases may lead to fixation of mutations in the genome.The platform was the first to solve and describe the 3D structure of this DNA repair enzyme. Publication
Molecular mechanism for interleukin-8 sorting. Interleukins are signal proteins that are secreted from various types of blood cells (first discovered from white blood cells, also known as leukocytes). Some interleukins are located in high concentrations in descrete compartments, which will cause a rapid signalling e.g. when cells are damaged. In this project, the role of several sequence motives in interleukin-8 we studied with respect to the sorting of interleukins to a special cellular compartment (WPB, Weibel-Palade Bodies). The facility assisted in model building and analysis of protein structures. Publication.
Structure of mutant sliding clamp - Sliding clamps are ring shaped proteins that encircle double stranded DNA. The clamps were first known for their role in DNA-replication as they link the DNA-polymerase to the DNA template to ensure a rapid and processive DNA synthesis. Sliding clamps interact with a variety of proteins and that they have an important role in other DNA metabolic processes, including DNA repair. In this project, a mutant version of the E. coli sliding clamp was studied with respect to initiation of DNA replication. The facility solved the 3D structure of the mutant clamp. Publication
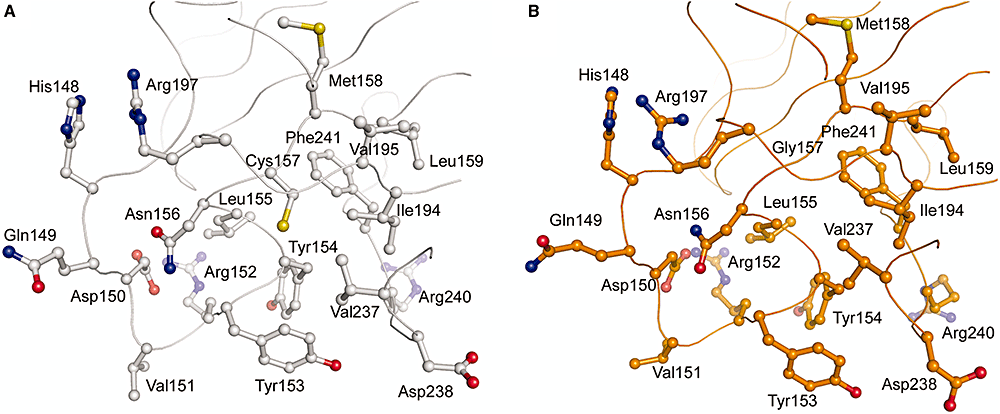
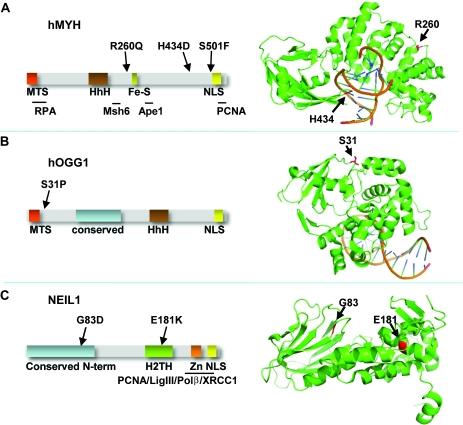
Separation-of-function mutants of a DNA glycosylase - The human DNA glycosylase hOGG1 is critical for removal of oxidized bases from DNA. The exact reaction mechanism for OGG1 has been debated with several concurrent models. THrough the design of separation-of-function mutants we were able to support the monofunctional hypothesis by biochemical and structural experiments. Publication
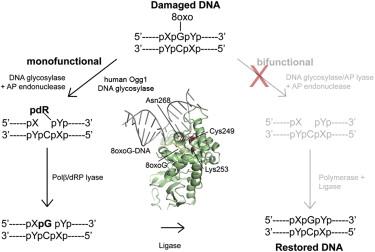
Analysis of mutant DNA glycosylases in patients with primary sclerosing cholangitis and cholangiocarcinoma - Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterized by inflammatory destruction of the bilary tree, and is often complicated by the development of cholangiocarcinoma (CCA). In this project, the genetic variation and possible reduced activity of several DNA repair enzymes were investigated for possible higher risk of CCA in PSC patients. The facility assisted with struvtural modeling and analysis of molecular basis for reduced enzymatic activity of several identified mutants. Publication