
Nucleotyping
The relation between chromatin organisation characteristics and cancer prognosis has been one of our main research projects since the establishment of the Institute in 2004. Chromatin is a complex of DNA and histones that are packaged into thin fibres within the nucleus of eukaryotic cells.
The packaging of DNA affects gene expression and regional mutation frequencies and contributes to carcinogenesis. In 2018, we presented a pan-cancer assay for automatic and objective detection of aberrant chromatin organisation by analysing the texture in images of DNA-specifically stained tumour cell nuclei (Lancet Oncology). Another related chromatin marker for gynaecological cancers was published in the Journal of the National Cancer Institute.
Nucleotyping identifies patient subgroups where this biomarker can be expected to greatly improve on current treatment guidelines with respect to the selection of candidates for adjuvant treatment after tumour removal. The aim of this project is to validate Nucleotyping in endometrial cancer and lung cancer.
Chromatin organisation and cancer
Each human cell contains about two metres of DNA. Most of the DNA is located in the nucleus, which is a membrane-enclosed organelle with a diameter of around six to ten μm. Specialised proteins, such as histones, bind to and fold the DNA into increasingly more condensed chromatin. Highly condensed chromatin is referred to as heterochromatin and contains genes in a transcriptionally repressed state, while weakly condensed chromatin, called euchromatin, contains genes that are accessible for transcription. Despite being tightly folded, the organisation is highly dynamic, and DNA can easily become available for replication, repair or transcription.
.gif)
Gene expression is regulated by mechanisms such as DNA methylation and histone modifications (Allis and Jenuwein, 2016). These and other heritable traits not coded in the DNA are referred to as epigenetics. After decades of research on genetics, e.g., through the Human Genome Project (International Human Genome Sequencing Consortium, 2004), epigenetics has received growing attention, e.g., through the Roadmap Epigenomics Project directed by the US National Institutes of Health (NIH) (Nature, 2015). Through efforts like these, epigenetics has been linked to disease pathogenesis by observations that higher-order chromatin organisation alters during cell differentiation (Dixon et al., 2015) and is the dominant determinant of variation in regional mutation rates in cancer cells (Polak et al., 2015; Schuster-Böckler and Lehner, 2012).
Chromatin organisation as a prognostic marker
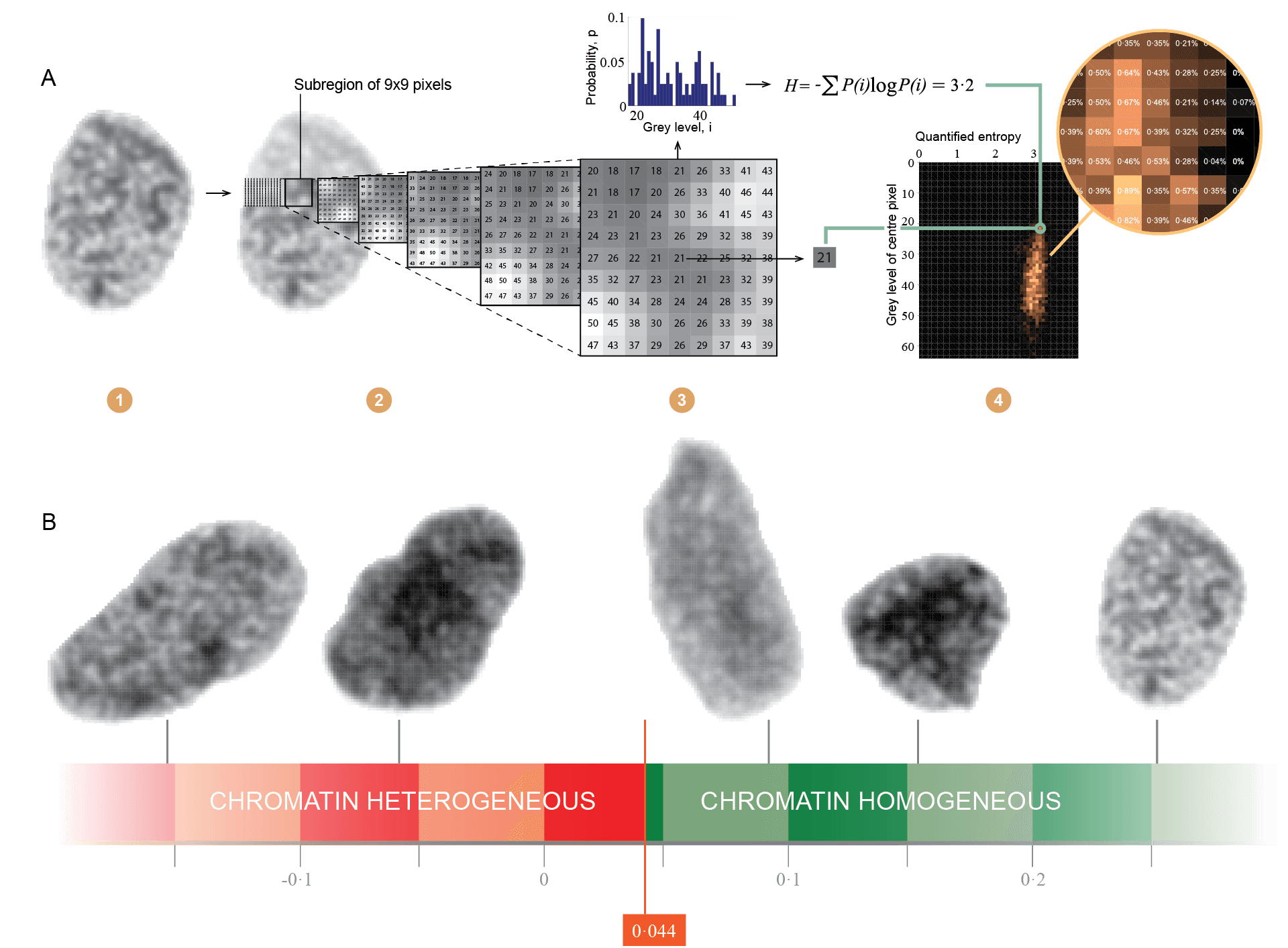
The higher-order organisation of chromatin can be studied by imaging DNA-specifically stained cell nuclei with a bright-field microscope, illustrating where highly condensed chromatin are visible as dark regions, and weakly condensed chromatin are visible as bright regions. By digitising such images and analysing the image texture, i.e., the spatial arrangement of pixels in an image, different aspects of the chromatin organisation in the imaged nuclei can be quantified using automatic computational methods. Estimating the entropy of chromatin structures, which relates to how disorganised or chaotic the chromatin appears, has been of particular interest in the collaboration between the Institute for Cancer Genetics and Informatics at Oslo University Hospital and the Department of Informatics at the University of Oslo, and the diagnostic and prognostic value of such objective markers have been evaluated in several human cancers (Yogesan et al., 1996; Jørgensen et al., 1996; Dunn et al., 2011; Nielsen et al., 2012, 2015, 2018; Hveem et al., 2017; Kleppe et al., 2018;).
In a recent study published in The Lancet Oncology (Vol 19, 2018), we developed a generic assay for automatic detection of aberrant chromatin organisation using a colorectal cancer patient series (see the animation on https://youtu.be/iwY3V98Y0V4). We independently validated the marker in six patient cohorts covering different cancer types, hospitals, time periods and technical image attributes. Patients found to have aberrant chromatin organisation, termed chromatin heterogeneous (CHE), consistently had shorter cancer-specific survival (CSS) compared to patients with chromatin homogeneous (CHO) tumours.
The aim of this project is to further develop, test and validate Nucleotyping as a prognostic biomarker. The Nucleotyping Project has three different goals organized as separate sub-projects:
- Validation of Nucleotyping in endometrial cancers
- Validation of Nucleotyping in lung cancers
- Adaptation of Nucleotyping to analyse nuclei in routine histological tissue sections
Validation of Nucleotyping in endometrial cancers
One of the analysed cohorts studied in The Lancet Oncology paper comprised 791 consenting endometrial cancer patients in the Molecular Markers in Treatment of Endometrial Cancer (MoMaTEC) trial. The 118 (15%) patients with CHE curettage specimen had shorter 5-year CSS when compared to CHO patients in univariate analysis (hazard ratio [HR] 4.4, 95% confidence interval [CI] 2.8-6.9; p<0.0001) and in multivariable analysis with the preoperatively available markers of age (continuous) and curettage histological risk classification (high- vs low-risk) (HR 1.9, 95% CI 1.1-3.1; p=0.013).
In FIGO stage I, 346 of the analysed patients belonged to the clinical low-risk group, 162 to intermediate-risk and 98 to high-risk. Only nine of the 508 patients at clinically low- or intermediate-risk died of endometrial cancer within five years, with no major survival difference observed between CHO and CHE patients. In the clinically high-risk group, 62 (63%) patients had CHO tumours, of whom only two died of endometrial cancer within five years, giving a 5-year CSS of 97%. The 36 (37%) CHE patients with FIGO stage I clinically high-risk had a 5-year CSS of 59%
The aim of this sub-project is to evaluate the clinical usefulness of the marker of chromatin heterogeneity in endometrial cancer. This automatic and objective marker was developed on colorectal cancer and successfully validated on several cancer types, including endometrial cancer using patients from the MoMaTEC trial. Patients with chromatin homogenous (CHO) tumours rarely died of endometrial cancer, whereas nearly all endometrial cancer-related deaths occurred in the chromatin heterogeneous (CHE) group. If these findings are replicated in several independent patient series, patients with CHO tumours could be spared adjuvant treatment, such as pelvic radiation or systemic chemotherapy, which cause significant long-term morbidities.
Prospective evaluation of retrospective cohorts is highly suitable to study this aim and is much faster and cheaper to perform than prospective, randomised trials and does not put patients at risk should the hypothesis be incorrect. Our partners will contribute with formalin-fixed paraffin-embedded (FFPE) hysterectomy specimens from approximately 800 patients treated at Oslo University Hospital, Norway, 150 treated at the Department of Gynaecology and Obstetrics, Innsbruck Medical University, Austria, and at least 100 patients treated at VU University Medical Center Amsterdam or the Academic Medical Center, The Netherlands.
These cohorts will facilitate a detailed analysis of the study aim. While only CSS data was available for analysis in the MoMaTEC study, the present study will also investigate time to and location of recurrences, which could reveal additional attributes of the marker, particularly in the low- and intermediate-risk groups where treatable vaginal or pelvic recurrences may occur without causing death. This will allow us to understand whether chromatin heterogeneity only identifies the most aggressive endometrial cancers that cause death or also identifies some less aggressive cancers that require additional treatment but will eventually be cured. We should also have sufficient data to reliably estimate the prognostic value in clinically relevant patient subgroups. Data on other prognostic factors will be available for some cohorts, in particular L1CAM data for the Oslo cohort, which we will utilise to investigate interactions with the chromatin heterogeneity marker with respect to clinical progression.
Based on the subgroup’s findings in the MoMaTEC study, we hypothesise that the 5-year CSS is about 95% for CHO patients with clinical high-risk FIGO stage I endometrial cancer and about 60% for the corresponding CHE patients. If this is true, then about 60% of patients in the clinical high-risk FIGO stage I group with CHO tumours can be spared some of the long-term morbidities associated with systemic chemotherapy, such as neuropathy (ref Lindemann) or gastro-intestinal dysfunction as with pelvic radiation (ref Vistad). However, the MoMaTEC validation study of the chromatin heterogeneity marker did not pre-plan any subgroup analyses; thus we need to confirm the subgroup findings in independent patient series. More patients could also allow a more comprehensive analysis of this subgroup, e.g., with respect to lymphovascular space invasion (LVSI) and histological type.
Cancer heterogeneity is a well-known phenomenon that is currently being actively researched, also in endometrial cancer (Mota et al., 2017; McAlpine et al., 2018). In order to study the implication of tumour heterogeneity with respect to the clinical utility of the chromatin heterogeneity marker, we wish to assess the intra-tumour variation of the marker and relate these observations to clinical progression. This will be done using the Oslo cohort, by including two samples from each hysterectomy specimen in addition to the initially analysed sample from each patient. In the present study, the two additional samples will only be used to analyse tumour heterogeneity; further studies would be needed if cancer heterogeneity should be deemed essential for the clinical application.
This study may potentially establish chromatin heterogeneity as a reproducible tumour feature that seemingly can guide adjuvant treatment decisions in endometrial cancer and thereby improve the risk-benefit ratio of adjuvant treatment. If so, we intend to use the evidence collected in this study to design prospective, randomised trials on adjuvant radiotherapy or chemotherapy in the patient subgroups where a substantial improvement of current clinical guidelines is necessary. We expect the clinical application of this marker to be cost-effective, as it is relatively simple and inexpensive compared to more modern approaches, such as genome sequencing.
The multivariate analysis will also include the biomarkers L1CAM and ProMisE (MSI, P53, and POLE) in addition to the features in standard clinical use.
Validation of Nucleotyping in lung cancers
Lung cancer is the leading cause of cancer mortality, resulting in around 2200 deaths in Norway each year. As the median age at death is around 70 years, almost as many life years are lost to lung cancer as to breast, colon and prostate cancer combined.2 Most lung cancers today are diagnosed in former or never-smokers, and about 50% of the cases are diagnosed in females. In patients below 60 years of age, more females than males are diagnosed with lung cancer. The fraction of patients diagnosed with early-stage disease is increasing, and currently, 20-25% undergo surgical treatment. A substantial fraction of surgically resected patients, almost one in two, recur and die from the disease.
About 85% of all lung cancer incidences are non-small cell lung cancers (NSCLCs).
Patients diagnosed with stage I NSCLC are recommended surgery, but are normally not prescribed adjuvant chemotherapy although it could be considered by a multidisciplinary tumour board as increased survival in some high-risk stage I subgroups has been suggested. In accordance with the recommendations of the European Society for Medical Oncology (ESMO), patients diagnosed with NSCLC with lymphatic spread (stage II and III) are routinely offered surgery followed by combination chemotherapy to reduce the risk of recurrence and cancer death. Patients are recommended MRI and PET-CT imaging examinations in order to rule out metastasis.
However, the selection criteria used today to select patients for adjuvant chemotherapy are suboptimal and are based on age and the morphologically defined extent of the disease. Over 30% of patients that are thought to have low recurrence risk, and are not offered adjuvant chemotherapy, still recur from the disease. It is also recognised that a fraction of those treated with toxic chemotherapy would be cured by surgery alone. Sadly, these patients will suffer from side-effects, and a minority even die due to the toxic effect of chemotherapy. Thus, there is a strong clinical need to identify factors that can improve the selection process for adjuvant chemotherapy.
A number of prognostic factors have been studied in lung cancer, but thus far, none are found to outperform the disease stage, due to, e.g., non-universality, high costs or limited improvement on selection. A clinically ideal biomarker should provide high discrimination power between high- and low-risks (i.e., a high degree of decision support), be universal, inexpensive and, if possible, automatic in order to avoid scoring variability. Nucleotyping has demonstrated these qualities in several other cancer types, and could be expected to perform similarly in lung cancer. It will be particularly interesting to assess whether chromatin heterogeneous patients, normally thought to be low-risk, have sufficiently increased risk of recurrence and cancer death to warrant adjuvant Chemotherapy. Another point of interest will be whether chromatin homogeneous patients that are normally thought to be at intermediate or high-risk have a sufficiently low recurrence and death rates to safely avoid adjuvant chemotherapy. Either of these findings would suggest that Nucleotyping enables more appropriate selection of candidates for chemotherapy after resection of lung cancer and if implemented in the clinic, may increase survival or reduce the morbidities and costs associated with lung cancer treatment, thus resulting in more optimal usage of limited health resources.
We hypothesise that Nucleotyping has the potential to objectively assist decision-making in the adjuvant treatment selection for lung cancer patients. Over the course of the project period, we will analyse the rate of local recurrences, metastases and deaths (overall and cancer-specific) in CHE stage I NSCLC to study if adjuvant treatment is indicated in this subgroup. Similar analyses will be performed in stage IIa, IIb, and IIIa NSCLC to investigate whether patients with CHO tumours could be spared adjuvant chemotherapy and its associated morbidities. Subgroup analysis of patients below the age of 70 and patients above the age of 70 are planned as only the younger patients are routinely offered adjuvant chemotherapy. The aim is to answer if and for whom Nucleotyping can improve current clinical practice with regards to the selection of candidates for adjuvant chemotherapy after lung cancer resection.
If this validation, designed as a prospective specimen collection, retrospective blinded evaluation (PRoBE), indicates that Nucleotyping should be utilised in the routine clinic, we intend to conduct prospective trials. The trials will be randomised based on adjuvant chemotherapy in the subgroups where a change in current treatment recommendations is indicated, with the long-term objective of enhancing the management of lung cancer patients by focusing adjuvant chemotherapy on those most at risk of recurrence and cancer death. This objective is consistent with the wider precision medicine agenda in oncology and will increase the quality of life and survival of individual patients while avoiding unnecessary expenses in the treatment of cancer.
Our collaborator in this project, Dr Odd Terje Brustugun, MD, will provide formalin-fixed paraffin-embedded (FFPE) lung cancer specimens from 984 consenting patients surgically resected at OUS from 2006 to 2018. Clinical and pathological characteristics are available for each patient, as well as detailed follow-up data with a median follow-up of more than five years and including records of local recurrences, metastases and deaths connected to the cause of death. The molecular marker PD-L1 and the mutational status markers EGFR and ALK have already been evaluated in this material, facilitating a study of the associations between these markers, chromatin heterogeneity and clinical outcome. This patient cohort will permit detailed analysis of the study aims, including reliable estimation of Nucleotyping’s prognostic value in clinically relevant patient subgroups.
Tumour heterogeneity is a well-known phenomenon that is currently being actively researched, also in lung cancer. In order to study the implication of tumour heterogeneity with respect to the clinical utility of Nucleotyping, we wish to assess the intra-tumour variation of the chromatin heterogeneity marker and relate these observations to patient outcome. This will be done by including all available tumour samples from each patient, totalling 3710 tissue samples for analysis.
The images acquired to assess chromatin heterogeneity can be directly used by in-house software to automatically estimate DNA ploidy and stroma fraction. Since this would not require any additional laboratory work and we recently demonstrated that these factors indicate colorectal cancer death, we will also perform these analyses and examine whether they provide information about the aggressiveness of lung tumours not captured by Nucleotyping and other markers.
Adaptation of Nucleotyping to analyse nuclei in routine histological tissue sections
The Nucleotyping method described above requires monolayers of isolated nuclei, a preparation technique that is not commonly practised at routine pathology laboratories. Although it is a straightforward lab technique that can be easily established in any laboratory, Nucleotyping would be a more attractive biomarker in routine use if it could be applied directly on routine histological sections. This would also open the path for a fully automated method, with even further reductions in cost and time.
More importantly, we postulate that the accuracy, and hence prognostic strength, could be further enhanced by the analysis of nuclei in sectioned tissue. As the current methods are analysing the chromatin in isolated nuclei, it is, in fact, analysing a 2D projection of all chromatin in a 3D nucleus. One might assume that the resolution of the chromatin analysis could be improved by analysing thin sections of the nucleus and that this increased resolution might increase the accuracy of the detection of chromatin heterogeneity, and hence improve the strength of the biomarker.
On the other hand, the DNA content, which is one of the features calculated when assessing the biomarker and an important feature for normalisation between samples, will undoubtedly be less accurately measured in sections compared to whole nuclei.
A correct estimation of the nuclei DNA content based on measurements of incomplete nuclei in sections is more likely in tissues with predominantly round epithelial nuclei (such as in prostate, breast, and lung) compared to tissues with more elongated nuclei (such as colon).
We will attempt to re-train, test and validate our machine learning algorithm using three prostate cancer cohorts, with a total of 2 600 tumour blocks from radical prostatectomies of 866 patients followed for more than ten years after surgery. Monolayer-based Nucleotyping has already been performed for all patients, and results will be compared, although the main result will be the new biomarker’s ability to prognosticate the outcome for these patients, measured as the time to recurrence.
We are currently evaluating different classical texture analysis methods in thin Feulgen-stained sections from prostate cancer patients. The plan is to evaluate methods that we have previously evaluated in monolayers and recently in 2D sections from colorectal cancer patients, particularly GLEM features and GLEM4D features. In addition, we are also considering including RGLEM features, fractal features and markers based on clustering GLEM features of nuclei, as well as possibly modified GLEM methods that ignore some of the nuclear periphery or bright regions within the nuclei. Analyses of chromatin compartments and exploiting the context of nuclei are less likely to be evaluated at this point, although it could be relevant later. However, we need to investigate how tumour heterogeneity affects the chromatin analyses, and how to best account for it (e.g., use the worst measurement or the measurement average).
If the developed markers appear to provide adequate prognostic information in the training subset, then one (initially, perhaps more later) will be selected for evaluation in the test subset. If the performance is reasonable in the test subset as well, then a modified normalisation method should be developed by empirical evaluation before the method is evaluated in a different cohort without re-training.