Sacral anomalies
Stormorken syndrome
Severe neurological diseases in children
Research projects
| Molecular characterization of encephalopathies with childhood onset |
| Novel genes causing primary cilia dysfunction in congenital brain malformations (Doriana Misceo's project group) |
| The only project not focusing on brain malformations is our work on Stormorken syndrome (OMIM 185070), which was initiated >20 years ago in collaboration with Prof. Helge Stormorken. We have documented that the mutation STIM1 p.R304W causes this syndrome, and we have established mouse lines expressing the mutated protein: Studying the effect of the Stormorken syndrome causing mutation STIM1 R304W in mouse models |
Studying the effect of the Stormorken syndrome causing mutation STIM1 R304W in mouse models
Repeated bleeding in a young boy was how the first patient came to Professor Helge Stormorken’s attention in 1974. Patients with Stormorken syndrome (OMIM 185070) exhibit mild bleeding tendency, thrombocytopathy, thrombocytopenia, mild anemia, splenic aplasia/functional asplenia, tubular aggregate myopathy, and miosis (Stormorken et al., 1985; Stormorken, 2002).
We have shown that the missense mutation STIM1 p.R304W causes the autosomal dominant Stormorken syndrome in four families (Misceo et al., 2014). In a parallel study, the same mutation was identified in two additional families (Nesin et al., 2014). The R304W substitution is the most common STIM1 mutation, so far identified in 18 patients with Stormorken syndrome in 13 families of different geographic origin.
STIM1 is an endoplasmic reticulum (ER) membrane protein that initiates store-operated Ca2+ entry (SOCE) into the cell when ER Ca2+ levels are low. ER Ca2+ store depletion activates STIM1 by releasing an intramolecular "clamp" formed between two coiled coil domains (CC1 and CC3) of the protein, enabling the C terminus to extend and interact with the plasma membrane protein Orai1. Orai1 is a Ca2+ release-activated calcium (CRAC) channel that enables Ca2+ influx into the cytoplasm.
In order to study the physiological consequences of the STIM1 R304W mutation, we established knock-in mice expressing this mutation. STIM1 R304W was lethal in the homozygous state, however, the few surviving homozygous mice and the heterozygotes presented with skeletal muscle degeneration (Figure 1; Gamage et al., 2018). The mice also presented with increased bone volume, as well as subgingival hair growth on the lower incisors (Figure 1; Gamage et al., 2020).
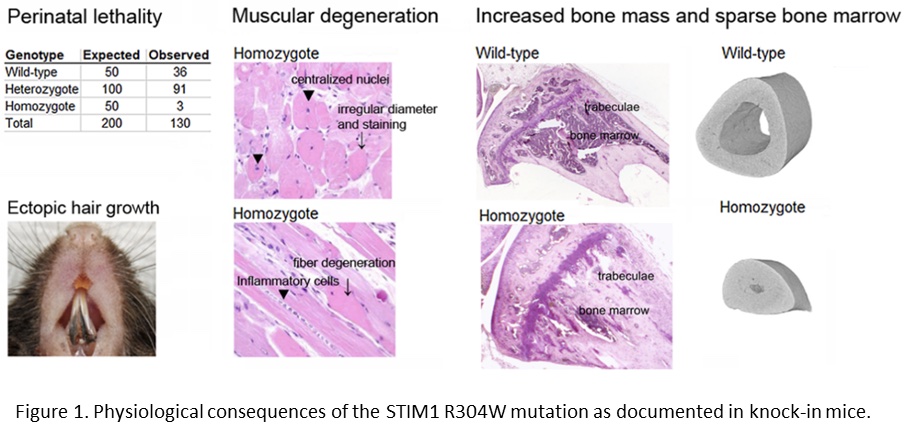
The findings in our mouse model highlight the importance of STIM1 in the development and homeostasis of the skeleton and the skeletal muscle. In mice, STIM1 R304W also seems to induce an abnormal epithelial cell fate.
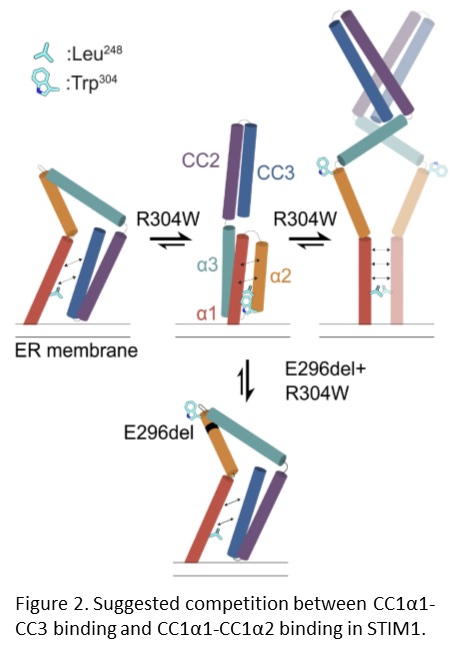
Our collaborators previously showed that the STIM1 R304W mutation destabilizes the resting state conformation of STIM1, resulting in a constitutively extended and therefore active STIM1, which leads to constitutive Ca2+ influx into the cell (Fahrner et al., 2018). We further documented that double mutant mice expressing STIM1 R304W and the deletion of Glu296 in cis, showed completely reversal of the pathophysiological effects of the STIM1 R304W mutation (Gamage et al., 2023). NMR spectroscopy, molecular dynamics simulations and live-cell experiments revealed that the deletion of Glu296 reversed the extended conformation of STIM1 R304W. Thus, the R304W and Glu296 double mutation restored the STIM1 CC domain interactions that are critical for proper regulation of the CRAC channel (Figure 2).
National collaborators:
Prof. G. Gunnes (NMBU): Pathology and histology
Dr. W. Louch and Prof. G. Christiansen (OUS): Ca2+ influx analyses
Profs. P.A. Holme and G. Tjønnfjord (OUS): Hematology
Profs. S.P. Lyngstadaas and J.E. Reseland (UiO): Analysis of bone structure
International collaborators:
Prof. C. Romanin (Johannes Kepler University Linz, Austria): STIM1 conformational changes
Prof. W. Bergmeier (University of North Carolina at Chapel Hill, USA): Mouse platelet biology
Prof. R. Lacruz (Sackler Institute, NYU School of Medicine, USA): Tooth development
Our papers in this project:
- Misceo et al. (2014). Human Mutation 35:556-564.
- Gamage et al. (2018). Cell Calcium 76:87-100.
- Gamage et al. (2020). Cell Calcium 85:102110.
- Gamage et al. (2023). Science Signaling 16(771):1-17.
Molecular characterization of encephalopathies with childhood onset
Severe encephalopathies in children are clinically heterogeneous, but life-long morbidity and increased mortality are common features. The genetic bases of these diseases are heterogeneous, and many patients remain without a molecular diagnosis. Our aim is to identify disease-causing variants in patients without a molecular diagnosis after diagnostic genetic tests, and to perform functional analyses to obtain knowledge about the biological consequences leading to the clinical presentation.
In this project, our main collaborator Prof. Petter Strømme (Division of Pediatrics and Adolescent Medicine, OUS/UiO) has included >100 patients from about 80 families negative for known lysosomal, mitochondrial, peroxisomal, and amino- and organic acidurias, and other relevant metabolic and degenerative diseases without history of infectious diseases or perinatal hypoxia.
We analyze Whole Genome or Exome Sequencing (WGS/WES) data in patient-parent trios. We share potential «novel disease genes» through GeneMatcher.org to identify unrelated patients with overlapping phenotypes and mutations affecting the same gene, which will support disease causality. We study patient cells in vitro and use animal models (such as zebrafish), to explore hypotheses about the molecular consequences of variants detected in «novel disease genes».
In patients remaining without a molecular diagnosis in our project, we further search for variants located in introns and untranslated regions (UTRs) and also for structural variants using long read-WGS and transcriptome analysis.
So far, we have detected the disease-causing gene variant in about 60% of the families analyzed (Table). This includes >20 novel mutations in previously described disease genes and several disease-causing variants in «novel disease genes». The mutations detected affect a range of cellular processes including RNA processing, regulation of gene expression, signal transduction, hormone transport, and the function of peroxisomes, lysosomes and the primary cilium.
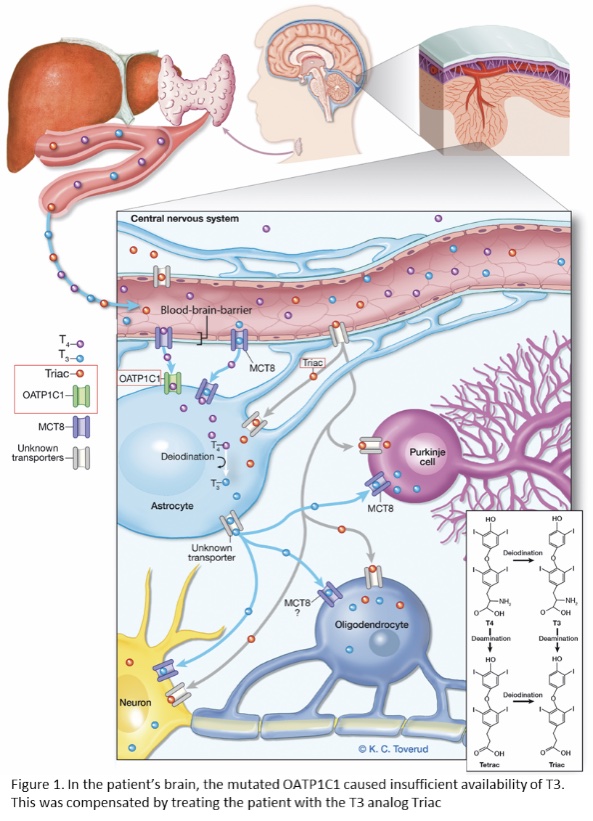
In six families, the genetic findings immediately led to a change in treatment options: e.g. diet adjustment, hematopoietic stem cell transplantation, or experimental medication. In one patient with a severe neurodegenerative disease, we identified a mutation in the brain specific T4 transporter protein OATP1C1. The progressive course of the disease appeared to be halted when the patient received experimental treatment with the T3 analog Triac (see Figure 1, Strømme et al., 2018). These results highlight the translational potential of our project.
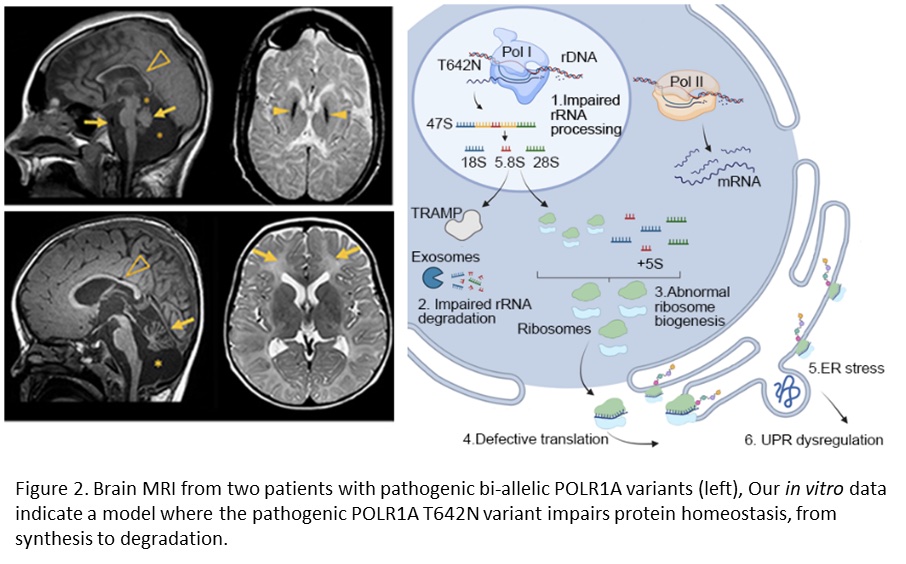
In a recent paper, we described two unrelated patients with hypomyelinating leukodystrophy, homozygous for POLR1A T642N. Studies in the patient’s fibroblasts documented that the mutated POLR1A led to aberrant rRNA processing and nucleolar morphology, causing abnormal protein homeostasis, and endoplasmic reticulum stress responses (see Figure 2, Misceo and Lirussi et al, 2023).
We have identified additional patients with pathogenic bi-allelic variants in POLR1A and we continue our studies of the molecular basis of the POLR1A related leukodystrophy.
WES, WGS, RNAseq, ChIPseq and PacBio sequencing are performed at the Norwegian High-Throughput Sequencing Centre (NSC), and the data storage and analyses performed at the National Infrastructure for High Performance Computing and Data Storage in Norway (Sigma2).
The project receives financial support by the Nasjonal kompetansetjeneste for sjeldne diagnoser (Norwegian National Advisory Unit on Rare Disorders, NKSD), Norway.
National collaborators
Prof. Strømme (Department of Pediatrics, OUS) and Dr. M. Fannemel (AMG): patient recruitment, counseling and clinical investigations.
Dr. Ying Sheng, Dr. Arvind Sundaram and Dr. Pål Marius Bjørnstad (AMG): bioinformatics analyses.
Dr. Lisa Lirussi (Ahus): in vitro work
Prof. H. Nilsen (OUS and Ahus): in vitro work.
Examples from this work:
- Misceo et al. (2023). A homozygous POLR1A variant causes leukodystrophy and affects protein homeostasis. Brain, in press.
- Sumathipala and Strømme et al. (2022). ZBTB11 dysfunction: spectrum of brain abnormalities, biochemical signature and cellular consequences. Brain 145:2602-2616.
- Epting et al. (2020). Loss of CBY1 results in a ciliopathy characterized by features of Joubert syndrome. Human Mutation 41:2179-2194.
- Sumathipala et al. (2019). TBCK encephaloneuropathy with abnormal lysosomal storage: use of a structural variant bioinformatics pipeline on WGS data unravels a 20-year-old clinical mystery. Pediatric Neurology 96:74-75.
- Strømme et al. (2018). Mutated thyroid hormone transporter OATP1C1 associates with severe brain hypometabolism and juvenile neurodegeneration. Thyroid 28:1406-1415.
- Kotlarz et al. (2018). Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nature Genetics 50:344-348.
- Barøy et al. (2015). A Novel Type of Rhizomelic Chondrodysplasia Punctata, RCDP5, Is Caused by Loss of the PEX5 Long Isoform. Human Molecular Genetics 24:5845-54.